本文共 2363 字,大约阅读时间需要 7 分钟。
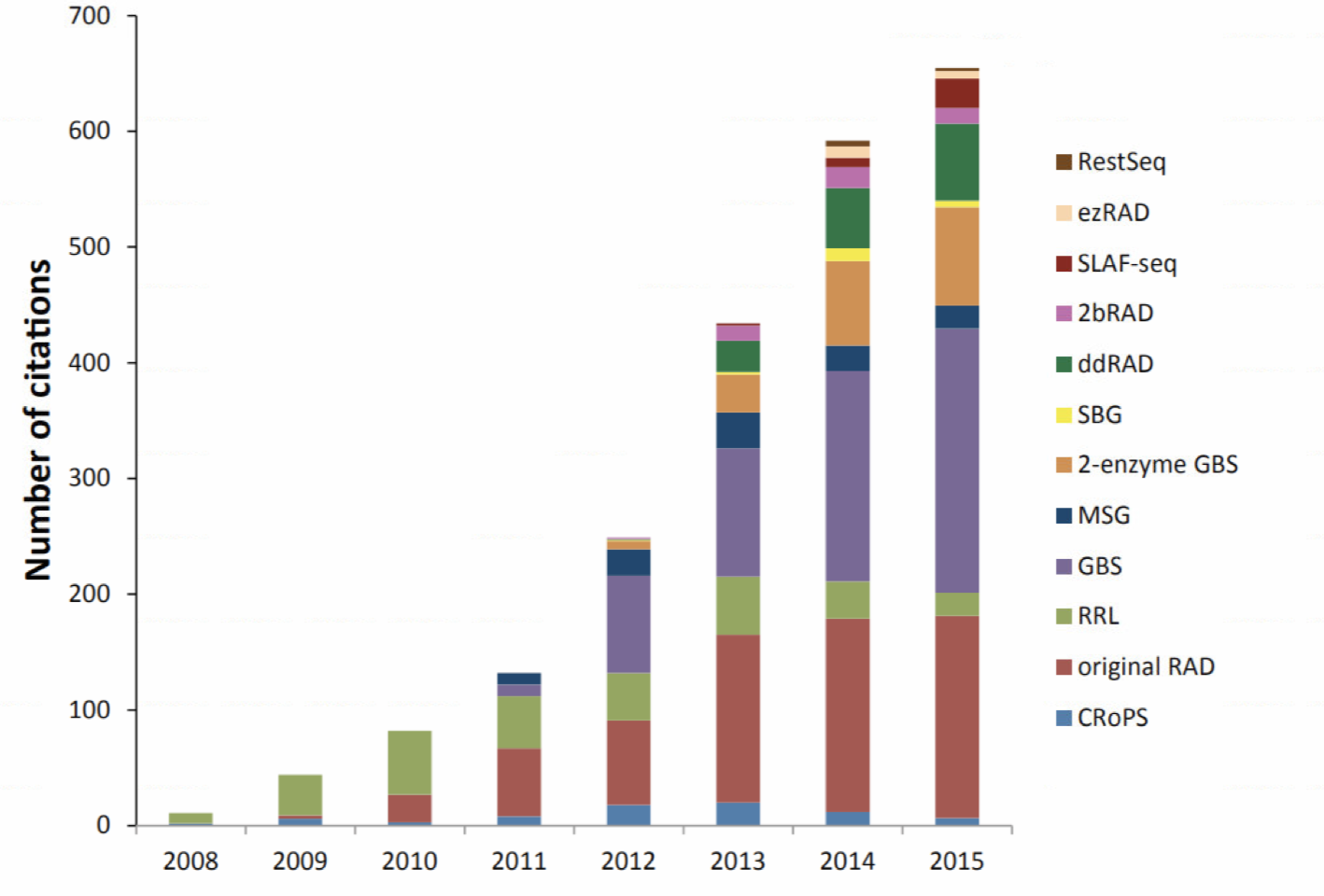
RAD-Seq(restriction site-associated DNA sequencing)最开始指的是2008年发表在PLOS ONE上“Rapid SNP discovery and genetic mapping using sequenced RAD markers"提出的方法,目前该文章的引用已经达到1200+,现在指代的是一系列基于限制性内切酶的测序技术。同样在概念上被引申的还有GBS(genotyping-by-sequencing),只不过GBS的名字不能让你直接把它和限制性内切酶联想起来.总之,如果现在公司给你推荐GBS或RAD-seq时,可能未必和你想的一样,你需要仔细问下他们的建库手段。毕竟手段不同,你的实验设计,操作和结果都会发生变化。这是RAD-seq相关方法的历年引用情况
RAD-seq虽说方法很多,但是文库构建流程大致如下,不同方法在其中某些步骤存在差异
- 起始基因组DNA量:能否允许降解FNA
- 限制性内切酶酶解:限制酶种类,数量
- 酶切位点结合接头:接头类型
- 酶解片段大小选择:直接选择,间接选择
- 添加barcode混池:视v接头而异
- 测序类型选择:单端,双端
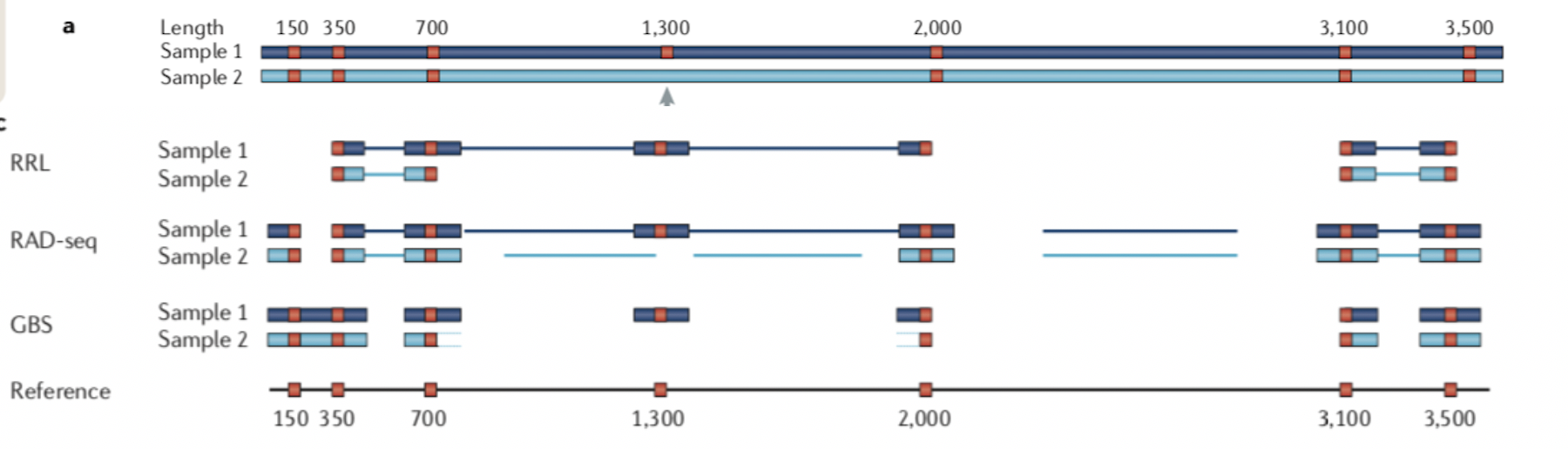
两者的差异在于,1)是现进行酶切然后随机破碎,最后仅选择存在酶切位点片段测序;2)也是酶切,但是后续直接选择合适大小的片段测序。
因此相对于1)测序的位点平均会少一点,也就会导致同一批样本后者利用率低于前者。无参考基因组更推荐前者,而不是后者。
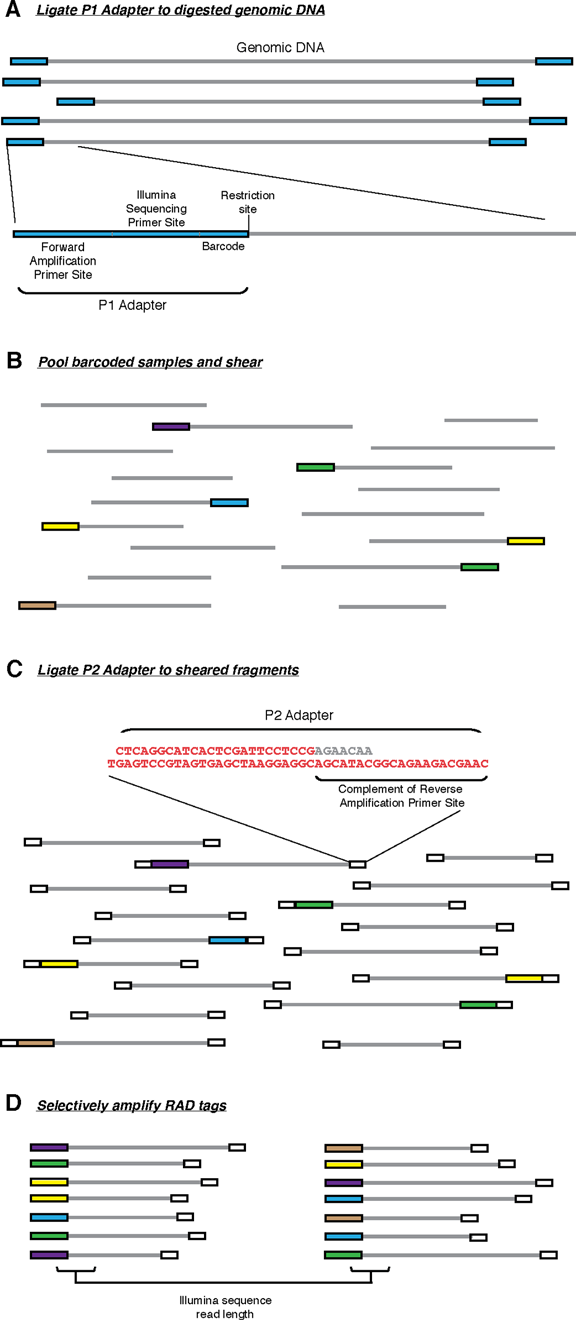
原始RAD-seqs
最先提出的RAD-seq技术流程,也就是RAD-seq的冠名技术,分为如下几步:
- 基因组DNA用限制性内切酶裂解, 然后连接到P1接头。P1接头里含有正向扩增和Illumina测序引物位点,以及4~5 bp 的核酸barcode. barcode至少大于3 bp。
- 之后接头连接的片段(adapter-ligated fragments)混池,随机打断
- DNA随后连接到P2接头,反向扩增扩展引物无法连接P2. P2是一种Y型接头,包含P2反向扩增引物位点的反向互补序列,使得不含P1接头的片段无法扩增。(Y型接头的工作原理)
- 最后仅有同时含P1和P2接头的片段能够上机测序。
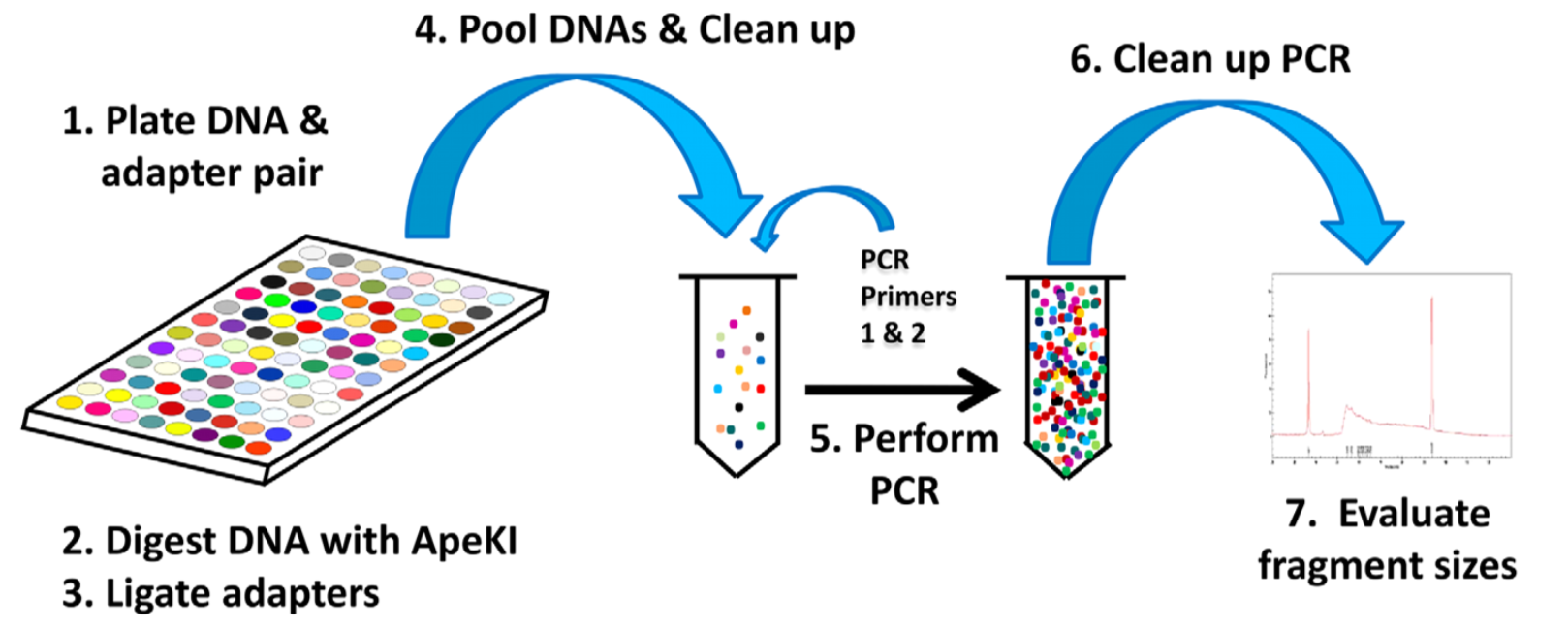
Genotyping-by-Sequencing
GBS比原始的RAD-seq步骤更加简单
- 将不同样本和含不同barcode接头成对放在平板里
- 使用ApeKI限制酶进行酶解
- 使用T4连接酶,将接头连接到片段两端因酶切产生的粘末端(stcky end)
- 将含不同barcode的样本混池,随后过片段长度筛选柱,过滤尚未反应的接头
- 加入PCR引物,进行PCR扩增
这里没有直接对片段进行筛选,但是PCR扩增时优先扩增小片段
ddRAD-seq
ddRAD-seq和GBS相似,两者都不需要在加接头后进行随机打碎,GBS通过PCR扩增的方式过滤了大片段,而ddRAD-seq通过双酶切的方式,然后筛选固定长度来选择合适大小的片段
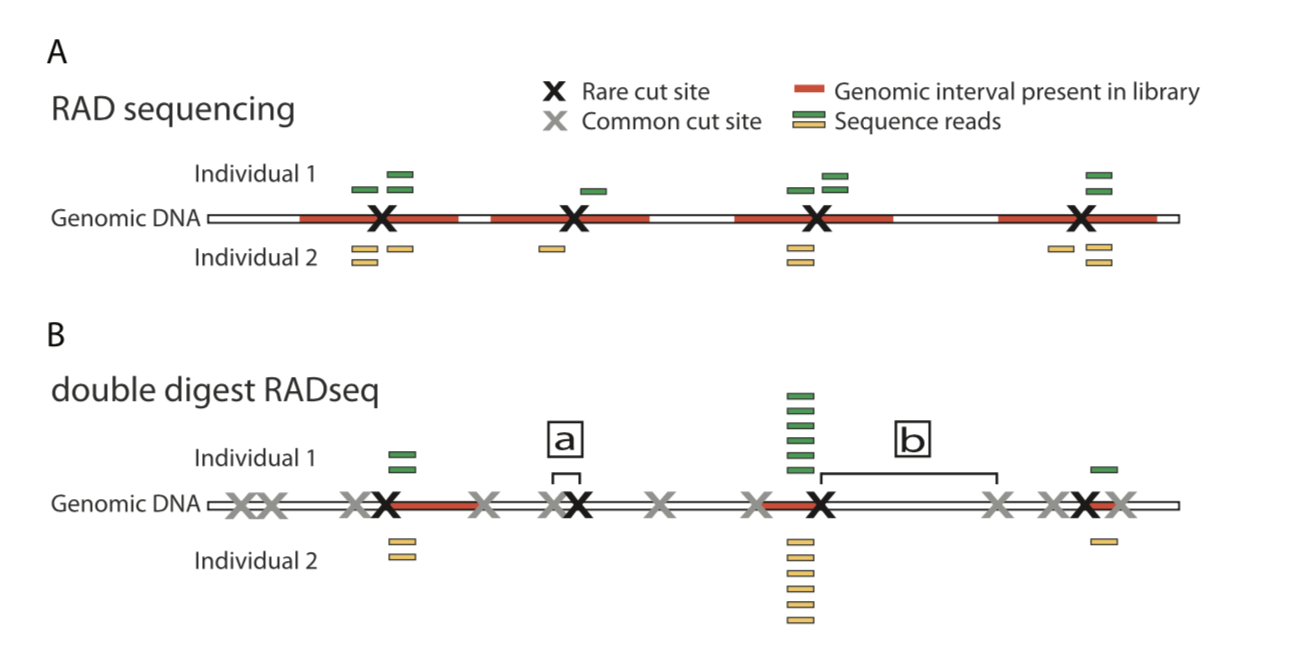
常见方法的比较
其实这些RAD-seq文库制备方法可以简单的分为两类:
- 1)对单酶切位点邻近片段测序,如最初的RAD-seq
- 2)对酶切位点两翼片段测序,如Genoytping-by-Sequencing
下面是常见的物RAD-seq方法比较
| 方法 | 原始RAD | 2bRAD | GBS | ddRAD | ezRAD |
|---|---|---|---|---|---|
| 控制位点的方法 | 选择限制酶 | 选择限制酶 | 选择限制酶 | 选择限制酶和片段大小选择阈值 | 选择限制酶和片段大小选择阈值 |
| 位点数/Mb | 30~500 | 50~1000 | 5~40 | 0.3~200 | 10~800 |
| 位点长度 | 300bp 或1kb contig | 33–36 bp | < 300 bp | < 300 bp | <300 bp |
| barcode费用/样本 | 低 | 低 | 低 | 低 | 高 |
| 添加barcode难度/样本 | 中等 | 低 | 低 | 低 | 高 |
| 是否用到专利试剂盒 | 否 | 否 | 否 | 否 | 是 |
| 识别PCR重复 | 使用双端测序 | 不能 | 使用降解的barcode | 用降解的barcode | 不能 |
| 特殊的设备 | 超声破碎仪 | 无 | 无 | Pippin Prep或普通的跑胶仪 | Pippin Prep或普通的跑胶仪 |
| 是否适用复杂和大基因组 | 好 | 差 | 中等 | 好 | 好 |
| 是否适用无参考基因组 | 好 | 差 | 中等 | 中等 | 中等 |
参考文献
- RAD-seq: Rapid SNP discovery and genetic mapping using sequenced RAD markers
- GBS: A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species
- ddRAD-seq: Double Digest RADseq: An Inexpensive Method for De Novo SNP Discovery and Genotyping in Model and Non-Model Species
- 2011 NATURE REVIEWS | GENETICS:Genome-wide genetic marker discovery and genotyping using next-generation
- 2016 NATURE REVIEWS | GENETICS:Harnessing the power of RADseq for ecological and evolutionary genomics
转载地址:http://mopxl.baihongyu.com/